
Advance/NanoLabo is a software for first-principles calculations and molecular dynamics, “designed for beginners.”
Advance/NanoLabo is a Graphical User Interface (GUI) designed to work with open-source materials analysis software, such as Quantum ESPRESSO and LAMMPS. You can search material databases such as Materials Project and easily set up modeling and computational conditions. First-principles and molecular dynamics calculations can be performed, and the results are visualized instantly. The intuitive and user-friendly GUI, designed for beginners, has garnered support from many users, with hundreds of sales both domestically and internationally since its launch in 2018.

Technical Specifications
Execution of Calculations
| Calculation Engine | Quantum ESPRESSO (First-Principles) LAMMPS (Molecular Dynamics) ThreeBodyTB (Genaral-Purpose Tight-Binding Method) |
| Calculation Functions | SCF Calculation, Structural Optimization, Hybrid Functional、vdW Correction, Band Structure, Density of States (w/ PDOS Calculator), Visualization of Charge Density, First-Principles MD, Car-Parrinello MD, Classical MD, MD with Neural Network Force Fields, Thermal Conductivity, Viscosity Coefficient, Diffusion Coefficient, Radial Distribution Function, TD-DFT, XAFS/EELS, Phonon (Effective Charge, Dielectric Constant, Band Structure, Density of States), NEB Method, Work Function (ESM Method) |
| Resources | Local Machine Calculation Server (Supports SSH Connection and Job Management with PBS/SLURM/PJM) Cloud (Mat3ra, Science Cloud GPU) NanoLabo Cloud Desktop |
modeling
| Materials Database | Materials Project PubChem |
| Crystal Systems | Cell Translation, Supercell, Impurity Substitution, Lattice Defects, Space Group Determination, Primitive Cell Transformation, Standard Cell Transformation |
| Surface and Interface Systems | Surfaces with Any Orientation, Molecular Adsorption on Surfaces, Mismatched Interfaces (Pro Version Only) |
| Molecular Systems | Drawing Organic Molecules, Filling Solvent Molecules, Polymer Models (Pro Version Only) |
System Requirements
| OS | Windows 10/11 (64bit) AlmaLinux 8 (64bit) macOS 13 or higher (ARM64) |
| Machine Spec (Recommended) | CPU : Intel Core i7 or higher Memory : 10 GB or more |
Cases
- Calculation of the adsorption of CO on Platinum (111) surface via Density Functional Theory
- Calculation of single-crystal silicon via Density Functional Theory
- Model configuration by packing molecules and molecular dynamics simulation of liquid
- Calculation of anisotropic Young’s modulus of single-crystal Ni via molecular dynamics simulation
- Calculation of ionic conductivity at 300 K of Li3OCl1-xBrx with M3GNet, a general force field based on graph neural networks
A picture is worth a thousand words;
introduce ease of use through videos.
Not just ease of use, but also putting cutting-edge technology into practice.
We always incorporate the latest cutting-edge technology into our product development and offer it to users. In particular, when it comes to Neural Network force field technology, we support various analyses by combining open-source Graph Neural Network force fields with our in-house developed Advance/NeuralMD.
1. Universal Graph Neural Network Force Fields
See Details
Structural optimization and molecular dynamics calculations using open-source Graph Neural Network force fields such as Open Catalyst, M3GNet, and CHGNet can be performed. Pre-trained models are available for all of them, allowing users to apply them universally to various systems without the need for users to optimize Neural Networks themselves.
- Calculation of ionic conductivity at 300 K of Li3OCl1-xBrx with M3GNet, a general force field based on graph neural networks
- Moleculer Dynamics Simulation of LiZnxMn2-xO4 using M3GNet
- Determining the Bulk Modulus of Ti-6Al-4V through Universal Neural Network Potentials
We also offer Graph Neural Network force field fine-tuning services.
2. Our in-house developed Advance/NeuralMD
See Details
Our in-house developed Neural Network force field software, Advance/NeuralMD, is available. It incorporates various state-of-the-art technologies, including the automatic generation of force fields using self-learning hybrid Monte Carlo methods and GPU acceleration enabling calculations of systems with up to 100,000 atoms.
3. General-Purpose Tight-Binding Method (ThreeBodyTB)
See Details
It is compatible with the open-source general-purpose tight-binding method software, ThreeBodyTB. It can be universally applied to inorganic materials containing 65 elements, and allows for simplified calculations of density of states and band structures.
4. Jupyter Interface for NanoLabo
See Details
It is a service that allows you to display and operate Jupyter Lab on the screen of Advance/NanoLabo. It transfers the structure data modeled in Advance/NanoLabo to a server running Jupyter Lab to generate Atoms objects of ASE.
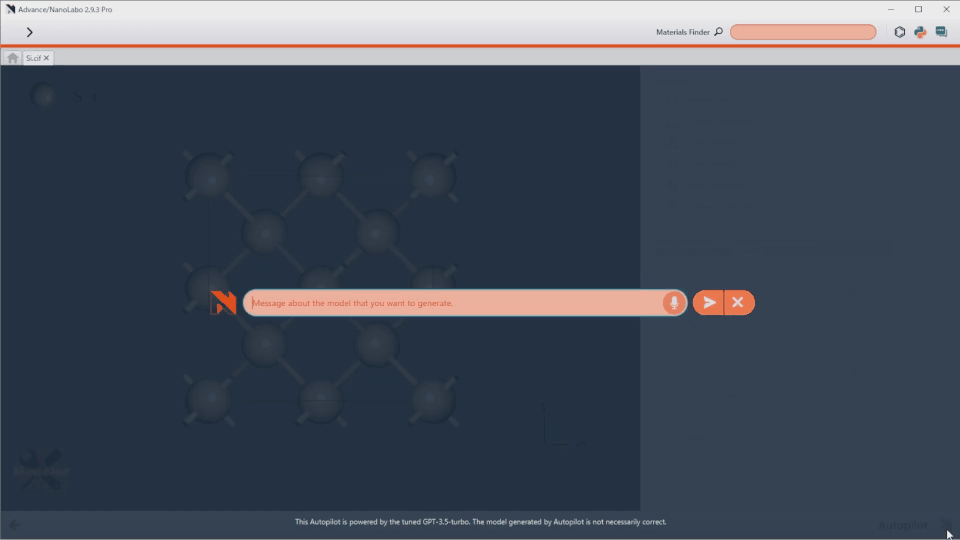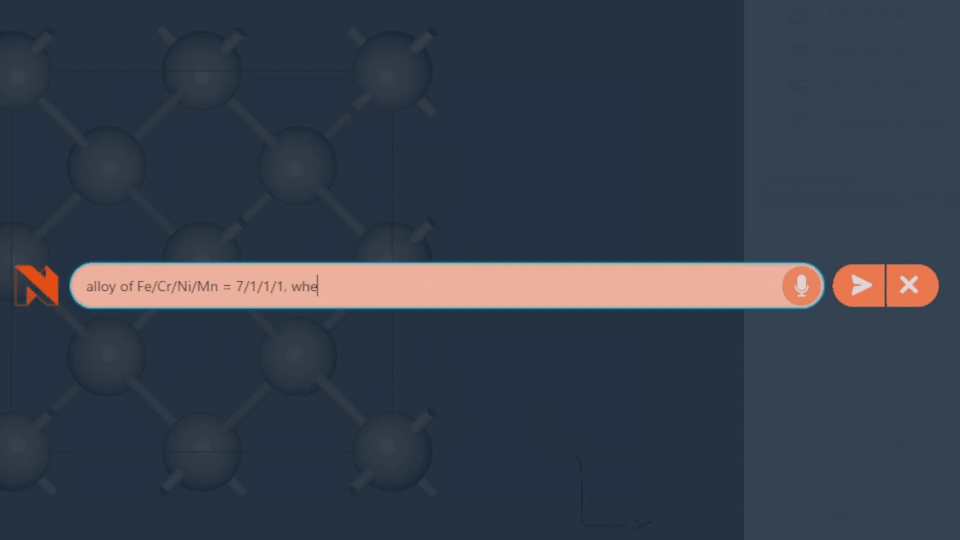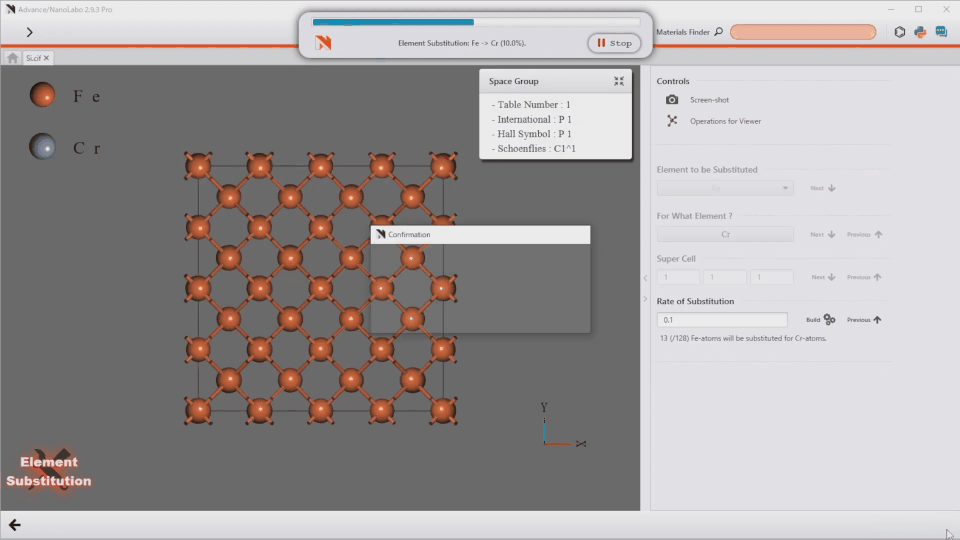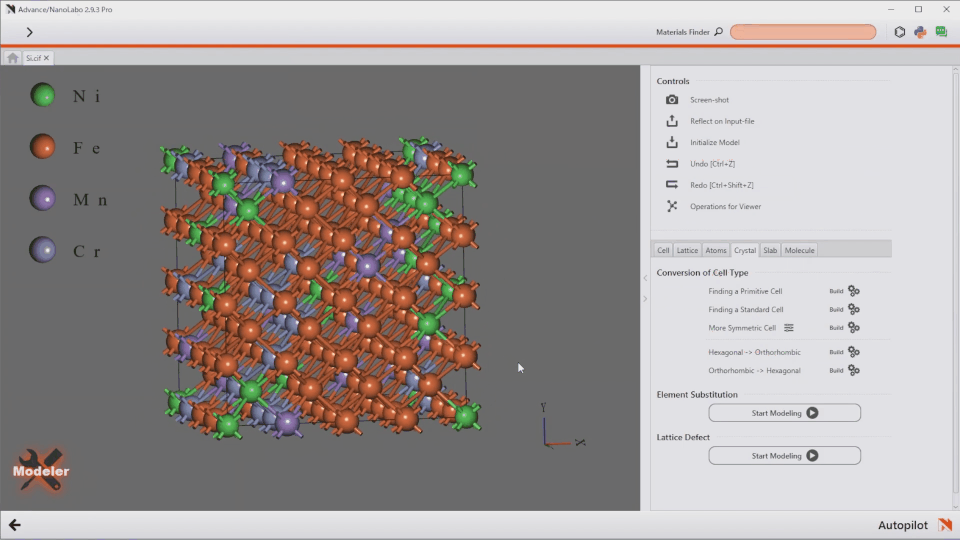
AI-Assisted Modeling
Introducing the world’s first Autopilot feature. Simply type your desired structure, and the system generates it instantly.
“Fe/Ni/Cr/Mn=7/1/1/1 alloy”
“ZrO2(001)/Rh(111) + 2 x NO@hollow”
Take full control of advanced modeling with flexible text input.

NanoLabo Cloud Desktop
Advance/NanoLabo is also available as a virtual desktop in the cloud. This service is provided in combination with workstation-level computing resources. The Python environment required to use graph neural network force fields is also pre-configured and ready to use. The cloud environment uses Microsoft Azure Virtual Desktop and is managed by AdvanceSoft.
Please see here for more details.
Try Advance/NanoLabo (1 Month Free)