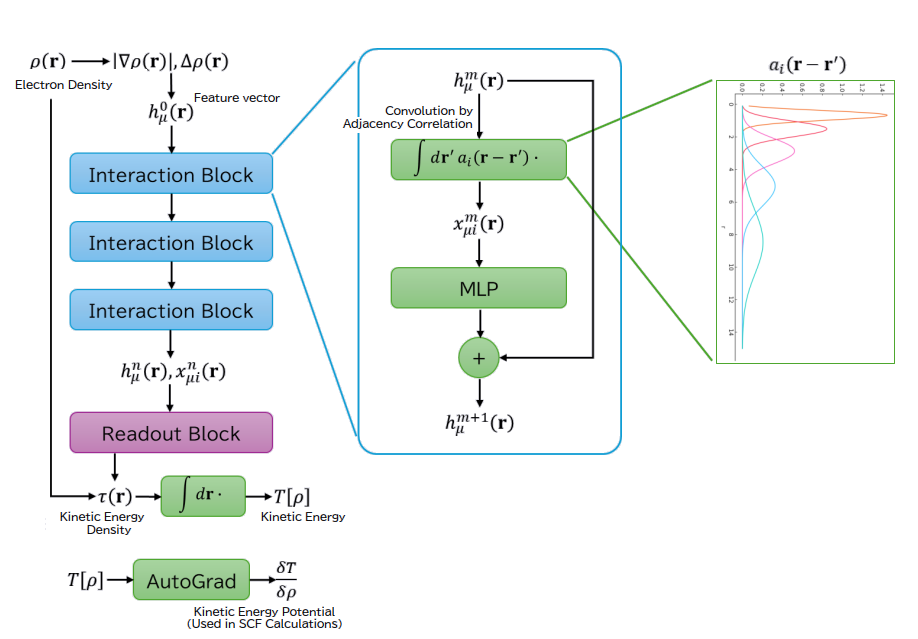
Deep Learned Orbital Free-DFT
Kohn-Sham DFT (KS-DFT) has long been the standard method in density functional theory (DFT). However, KS-DFT is computationally expensive and challenging for simulating large systems due to its reliance on orbital (wave function) representation. Orbital Free-DFT (OF-DFT), which directly optimizes electron density without using orbitals, offers extremely fast simulations at a much lower computational cost. The main challenge with OF-DFT has been the lack of a practical kinetic energy functional. AdvanceSoft Corporation has addressed this issue by applying its proprietary field deepening algorithm. We now offer services using our new product, Advance/OF-DFT, which incorporates the deep-learned kinetic energy functional AdvanceSoft25.
Kohn Sham-DFT vs Orbital Free-DFT
| KS-DFT | OF-DFT | Force Field | |
|---|---|---|---|
| Electron Density | available | available | not available |
| Orbital (Wave Variables) | available | not available | not available |
| Kinetic Energy | Explicitly calculated in orbital | Deep Learned Functional : AdvanceSoft25 | – |
| Calculation Accuracy | High | Depends on the functional | Depends on force field |
| Calculation Cost | 𝑂(𝑁3) | 𝑂(𝑁) | 𝑂(𝑁) |
| Versatility | Applicable to all elements | Pseudo-potentials should be expanded (to be resolved in next version) | Ensure versatility with GNNP |
Analyzing electronic structure, but same speed as GNNP
- The computational cost of OF-DF is proportional to the number of atoms (𝑁), denoted as (𝑂(𝑁)), so simulations can be performed at the same level of computational speed as Graph Neural Network Potential (GNNP).
- Since information on electron density is retained, Bader charge can also be analyzed after convergence of the SCF calculation.
- Doping of electrons and holes is possible, and in combination with the Effective Screening Medium (ESM), large-scale MD simulations with controlled electrode potentials can be realized (ESM is not implemented in the current version). Application of an external electric field is also easy.
- The type of exchange-correlation functional can be selected at the time of SCF calculation, so non-local correlations corresponding to dispersion forces such as vdW-DF and rVV can also be used depending on the system; empirical functions such as DFT-D3 are not required.
- Since the wavefunction information is not included, a separate KS-DFT calculation is required for band structure and density of states calculations.
Architecture of the deep learned
kinetic energy functional: AdvanceSoft25